SPACER Predict Notebook
Example Data Setup
Run the cell below to use the synthetic example dataset. Swap the three path variables for your own files when running on real data.
Variable |
Example value |
Description |
|---|---|---|
|
|
377-gene reference list |
|
|
600-cell AnnData (400 tumour + 200 stromal) |
|
|
Model saved by |
The preprocessing hyper-parameters (RADIUS, RESOLUTION, N_GENES) must match the values used in ``train.ipynb``, otherwise the gene panel and bag construction will not line up with the trained model.
Synthetic label layout: Macrophage+ cells occupy the right half of the tissue (X > 190).
[ ]:
# ── Example data paths (swap for your own files) ──────────────────────────────
# Run `python create_example_data.py` once to generate the files below.
import os
GENE_CSV = 'data/example_genes.csv' # reference gene list
ADATA_H5AD = 'data/example_spatial.h5ad' # synthetic AnnData
MODEL_PATH = 'sample_output/best_model.pth' # produced by train.ipynb
IMMUNE_CELL = 'macrophage'
# ── Preprocessing hyper-parameters ───────────────────────────────────────────
# IMPORTANT: these MUST match the values used in train.ipynb, otherwise the
# selected gene panel / bag construction will not match the trained model.
RADIUS = 50 # neighbourhood radius used to build each bag
RESOLUTION = 'low' # 'low' | 'high'
N_GENES = 500 # number of top-expressed tumour genes to keep
# If train.ipynb has not been run yet, save an untrained placeholder
if not os.path.exists(MODEL_PATH):
import torch, pandas as pd
from model.model import MIL as _MIL
_genes = pd.read_csv(GENE_CSV)['Gene'].tolist()
os.makedirs(os.path.dirname(MODEL_PATH), exist_ok=True)
torch.save(_MIL(_genes).state_dict(), MODEL_PATH)
print(f"Saved untrained placeholder → {MODEL_PATH}")
print("Run train.ipynb first for a properly trained model.")
else:
print(f"Model found: {MODEL_PATH}")
[11]:
import csv
import torch
import numpy as np
import pandas as pd
from tqdm import tqdm
import scanpy as sc
from torch.utils.data import DataLoader
from model.model import MIL
[12]:
from torch.utils.data import Dataset
import scipy.sparse as sp
from scipy.spatial.distance import cdist
from tqdm import trange
from scipy.sparse import issparse
def map_immune_cell(tag: str) -> str:
mapping = {
"tcell": "T",
"bcell": "B",
"macrophage": "Macrophage",
"neutrophil": "Neutrophil",
"fibroblast": "Fibroblast",
"endothelial": "Endothelial",
}
if tag in mapping:
return mapping[tag]
raise ValueError(f"Invalid immune cell type: {tag}")
def preprocess_data(adata, immune_cell, n_genes=500, resolution="low"):
has_label = immune_cell is not None
if has_label and immune_cell not in adata.obs.columns:
immune_cell = map_immune_cell(immune_cell)
adata = adata.copy()
adata.var_names_make_unique()
tumour = adata[adata.obs["cell_type"].astype(int) == 1].copy()
if issparse(tumour.X):
mean_expr = np.asarray(tumour.X.mean(axis=0)).ravel()
else:
mean_expr = tumour.X.mean(axis=0)
gene_names = tumour.var_names
n_genes = min(n_genes, int(0.2 * len(gene_names))) if n_genes > len(gene_names) else n_genes
top_idx = mean_expr.argsort()[-n_genes:][::-1]
top_genes = gene_names[top_idx]
tumour_gene_set = [
"PRAME", "MUC1", "EPCAM", "PMEL", "MAGEA3", "WT1",
"MYC", "CCND1", "CDK4", "CDK6", "BCL2", "BIRC5",
]
hla_genes = [g for g in adata.var_names if g.startswith("HLA")]
keep = list(dict.fromkeys(tumour_gene_set + hla_genes + list(top_genes)))
drop = {"CD68", "STAT1", "MMP13"}
keep = [g for g in keep if g in adata.var_names and g not in drop]
adata = adata[:, keep].copy()
if has_label:
adata.obs[immune_cell] = adata.obs[immune_cell].astype(float)
if resolution != "high":
if not set(adata.obs[immune_cell].unique()).issubset({0, 1}):
thresh = np.percentile(
adata[adata.obs["cell_type"] == 1].obs[immune_cell], 75
)
adata.obs[immune_cell] = (adata.obs[immune_cell] > thresh).astype(int)
else:
adata.obs["dummy_label"] = -1
immune_cell = "dummy_label"
return adata
class BagsDataset(Dataset):
# mode='train': returns [pos + k-1 neg] bags; mode='infer': returns one bag (label=-1)
def __init__(self, input_data, immune_cell="tcell", max_instances=None,
radius=200, resolution="low", n_genes=500, k=2, mode="train"):
self.mode = mode.lower()
self.k = k
self.radius = radius
self.resolution = resolution
self.max_instances = max_instances
self.n_genes = n_genes
self.immune_cell = (
map_immune_cell(immune_cell) if (immune_cell and self.mode == "train") else None
)
if isinstance(input_data, str):
df = pd.read_csv(input_data)
adata_list = []
for _, row in df.iterrows():
ad = sc.read_h5ad(row["adata"])
r = row.get("radius", self.radius)
res = row.get("resolution", self.resolution)
ad_prep = preprocess_data(ad, self.immune_cell, n_genes, res)
adata_list.append((ad_prep, r, res))
self.batches = self._build_bags(adata_list)
elif hasattr(input_data, "X"):
ad = preprocess_data(input_data, self.immune_cell, n_genes, resolution)
self.batches = self._build_bags([(ad, radius, resolution)])
else:
raise ValueError("input_data must be an AnnData or a CSV path")
def __len__(self): return len(self.batches)
def __getitem__(self, idx): return self.batches[idx]
def _build_bags(self, adata_radius_list):
train_batches, infer_items = [], []
for adata, radius, resolution in adata_radius_list:
coords = adata.obs[["X", "Y"]].astype(float).to_numpy()
expr = adata.X
barcodes = adata.obs_names.to_numpy()
gene_names = adata.var_names.tolist()
if self.mode == "train":
labels = adata.obs[self.immune_cell].astype(int).to_numpy()
else:
labels = np.full(adata.n_obs, -1, dtype=int)
cell_types = adata.obs["cell_type"].astype(int).to_numpy()
pos_bags, neg_bags = [], []
for i in trange(adata.n_obs, desc=f"[{self.mode}] r={radius}", leave=False):
if cell_types[i] == 0:
continue
dist = cdist([coords[i]], coords)[0]
neigh = np.where(dist <= radius)[0]
neigh = neigh[cell_types[neigh] == 1]
if resolution == "high":
neigh = neigh[neigh != i]
if len(neigh) == 0:
continue
if self.max_instances and len(neigh) > self.max_instances:
continue
bag = {
"distances": dist[neigh, None].astype(np.float32),
"gene_expression": expr[neigh],
"label": labels[i],
"core_idx": i,
"gene_names": gene_names,
"cell_id": barcodes[i],
}
if self.mode == "train":
(pos_bags if labels[i] == 1 else neg_bags).append(bag)
else:
infer_items.append(bag)
if self.mode == "train":
k_neg = self.k - 1
n_batch = min(len(pos_bags), len(neg_bags) // k_neg) if k_neg else len(pos_bags)
if n_batch == 0:
continue
np.random.shuffle(neg_bags)
pos_bags = pos_bags[:n_batch]
neg_bags = neg_bags[:n_batch * k_neg]
for b in range(n_batch):
train_batches.append(
[pos_bags[b]] + neg_bags[b * k_neg : (b + 1) * k_neg]
)
return train_batches if self.mode == "train" else infer_items
def custom_collate_fn(batch):
bags = batch[0] if isinstance(batch[0], list) else batch
dists, exprs, labels, cores, gnames, cids = [], [], [], [], [], []
for bag in bags:
dists.append(torch.tensor(bag["distances"], dtype=torch.float32))
ge = bag["gene_expression"]
if sp.issparse(ge):
ge = ge.todense()
exprs.append(torch.tensor(ge, dtype=torch.float32))
labels.append(torch.tensor(bag["label"], dtype=torch.float32))
cores.append(bag["core_idx"])
gnames.append(bag["gene_names"])
cids.append(bag["cell_id"])
return dists, exprs, labels, cores, gnames, cids
[13]:
device = torch.device("cuda" if torch.cuda.is_available() else "cpu")
print(f"Using device: {device}")
Using device: cpu
[14]:
def load_all_genes(reference_gene_file):
all_genes = []
with open(reference_gene_file, 'r') as csvfile:
reader = csv.DictReader(csvfile)
for row in reader:
all_genes.append(row['Gene'])
return all_genes
all_genes = load_all_genes(GENE_CSV)
print(f"Loaded {len(all_genes)} reference genes from {GENE_CSV}")
Loaded 377 reference genes from data/example_genes.csv
[15]:
model = MIL(all_genes).to(device)
[16]:
model.load_state_dict(torch.load(MODEL_PATH, map_location=device))
model.eval()
print(f"Loaded weights from {MODEL_PATH}")
Loaded weights from sample_output/best_model.pth
[ ]:
adata = sc.read_h5ad(ADATA_H5AD)
# radius / n_genes / resolution are passed through explicitly and MUST match
# the values used when the model was trained (see train.ipynb).
prediction_dataset = BagsDataset(
adata,
immune_cell=IMMUNE_CELL,
radius=RADIUS,
n_genes=N_GENES,
resolution=RESOLUTION,
mode='infer',
)
prediction_dataloader = DataLoader(prediction_dataset, batch_size=1, collate_fn=custom_collate_fn)
adata.obs['Macrophage_pred'] = np.nan
print(f"Prediction bags: {len(prediction_dataset)}")
[ ]:
def predict(model, prediction_dataloader, adata, device=None):
if device is None:
device = next(model.parameters()).device
model.eval()
with torch.no_grad():
for batch_data in tqdm(prediction_dataloader, desc="Predicting"):
# custom_collate_fn returns SIX objects, so unpack all six here:
# distances, gene_expressions, labels, core_idxs, gene_names, cell_ids
# labels and core_idxs are unused at inference time, hence the `_`.
distances_list, gene_expressions_list, _, _, gene_names_list, cell_ids_list = batch_data
distances_list = [d.to(device) for d in distances_list]
gene_expressions_list = [g.to(device) for g in gene_expressions_list]
outputs = model(distances_list, gene_expressions_list, gene_names_list)
if outputs is None:
continue
for output, cell_id in zip(outputs, cell_ids_list):
adata.obs.at[cell_id, 'Macrophage_pred'] = output.cpu().item()
[19]:
predict(model, prediction_dataloader, adata)
Predicting: 100%|██████████| 400/400 [00:00<00:00, 3273.39it/s]
[20]:
print(f"Total cells: {len(adata.obs)}")
print(f"Cells with prediction: {adata.obs['Macrophage_pred'].notna().sum()}")
adata.obs[['X','Y','cell_type','Macrophage','Macrophage_pred']].dropna().head(10)
Total cells: 600
Cells with prediction: 400
[20]:
| X | Y | cell_type | Macrophage | Macrophage_pred | |
|---|---|---|---|---|---|
| CELL_00000-1 | 0.0 | 0.0 | 1 | 0 | 0.225890 |
| CELL_00001-1 | 20.0 | 0.0 | 1 | 0 | 0.188221 |
| CELL_00002-1 | 40.0 | 0.0 | 1 | 0 | 0.139861 |
| CELL_00003-1 | 60.0 | 0.0 | 1 | 0 | 0.118036 |
| CELL_00004-1 | 80.0 | 0.0 | 1 | 0 | 0.113145 |
| CELL_00005-1 | 100.0 | 0.0 | 1 | 0 | 0.123089 |
| CELL_00006-1 | 120.0 | 0.0 | 1 | 0 | 0.167911 |
| CELL_00007-1 | 140.0 | 0.0 | 1 | 0 | 0.174974 |
| CELL_00008-1 | 160.0 | 0.0 | 1 | 0 | 0.158479 |
| CELL_00009-1 | 180.0 | 0.0 | 1 | 0 | 0.120093 |
[21]:
mask = ~adata.obs['Macrophage_pred'].isna()
adata = adata[mask].copy()
print(f"Cells after filtering to predicted: {len(adata.obs)}")
Cells after filtering to predicted: 400
[22]:
adata.obs['Macrophage'].value_counts()
[22]:
Macrophage
0 220
1 180
Name: count, dtype: int64
[23]:
adata.obs['Macrophage_pred'] = adata.obs['Macrophage_pred'].fillna(0)
[24]:
from sklearn.metrics import roc_auc_score
auroc = roc_auc_score(adata.obs['Macrophage'], adata.obs['Macrophage_pred'])
print(f"AUROC: {auroc:.4f}")
AUROC: 0.9826
[25]:
import matplotlib.pyplot as plt
import seaborn as sns
df = adata.obs.dropna(subset=['Macrophage_pred', 'Macrophage']).copy()
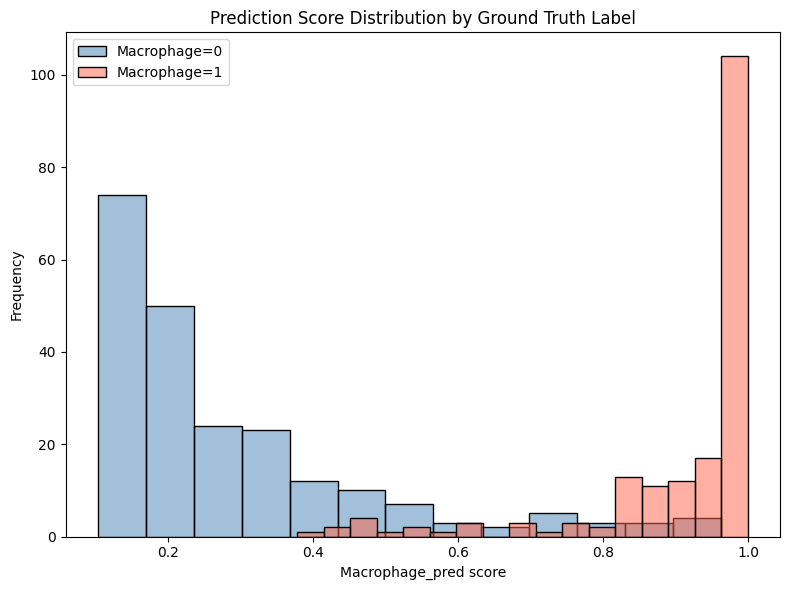
plt.figure(figsize=(8, 6))
sns.histplot(df.loc[df['Macrophage'] == 0, 'Macrophage_pred'],
color='steelblue', alpha=0.5, label='Macrophage=0')
sns.histplot(df.loc[df['Macrophage'] == 1, 'Macrophage_pred'],
color='tomato', alpha=0.5, label='Macrophage=1')
plt.legend()
plt.xlabel('Macrophage_pred score')
plt.ylabel('Frequency')
plt.title('Prediction Score Distribution by Ground Truth Label')
plt.tight_layout()
plt.show()
[26]:
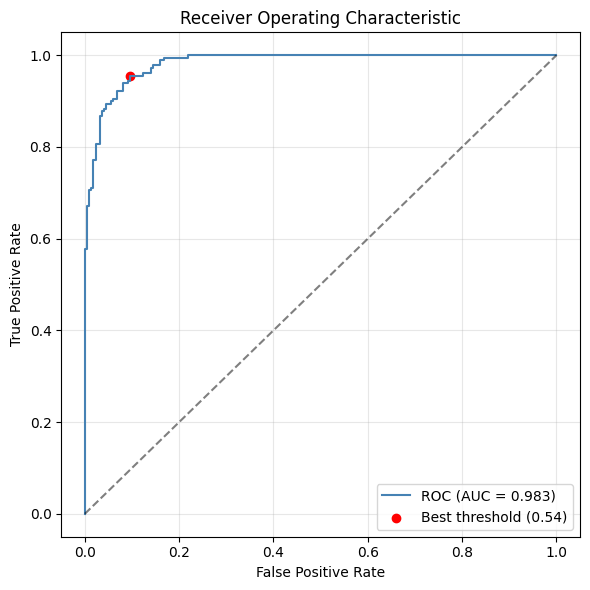
from sklearn.metrics import roc_curve, auc
df_roc = adata.obs.dropna(subset=['Macrophage_pred', 'Macrophage']).copy()
df_roc['Macrophage'] = df_roc['Macrophage'].astype(int)
fpr, tpr, thresholds = roc_curve(df_roc['Macrophage'], df_roc['Macrophage_pred'])
roc_auc = auc(fpr, tpr)
youdenJ = tpr - fpr
best_thresh = thresholds[np.argmax(youdenJ)]
print(f"Best threshold (Youden\'s J): {best_thresh:.3f} J={youdenJ.max():.3f}")
plt.figure(figsize=(6, 6))
plt.plot(fpr, tpr, label=f'ROC (AUC = {roc_auc:.3f})', color='steelblue')
plt.scatter(fpr[np.argmax(youdenJ)], tpr[np.argmax(youdenJ)],
marker='o', color='red', label=f'Best threshold ({best_thresh:.2f})')
plt.plot([0, 1], [0, 1], 'k--', alpha=0.5)
plt.xlabel('False Positive Rate')
plt.ylabel('True Positive Rate')
plt.title('Receiver Operating Characteristic')
plt.legend(loc='lower right')
plt.grid(True, alpha=0.3)
plt.tight_layout()
plt.show()
Best threshold (Youden's J): 0.539 J=0.860
[27]:
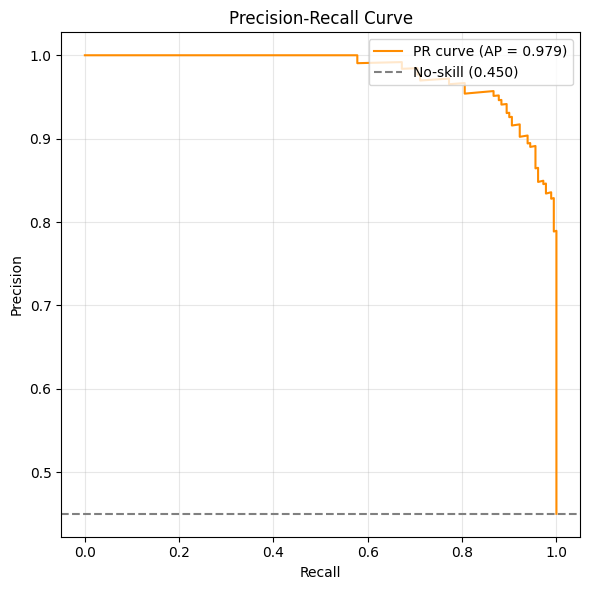
from sklearn.metrics import precision_recall_curve, average_precision_score
df_pr = adata.obs.dropna(subset=['Macrophage_pred', 'Macrophage']).copy()
df_pr['Macrophage'] = df_pr['Macrophage'].astype(int)
precision, recall, _ = precision_recall_curve(df_pr['Macrophage'], df_pr['Macrophage_pred'])
ap = average_precision_score(df_pr['Macrophage'], df_pr['Macrophage_pred'])
baseline = df_pr['Macrophage'].mean()
plt.figure(figsize=(6, 6))
plt.plot(recall, precision, color='darkorange', label=f'PR curve (AP = {ap:.3f})')
plt.axhline(y=baseline, color='gray', linestyle='--', label=f'No-skill ({baseline:.3f})')
plt.xlabel('Recall')
plt.ylabel('Precision')
plt.title('Precision-Recall Curve')
plt.legend(loc='upper right')
plt.grid(True, alpha=0.3)
plt.tight_layout()
plt.show()
[28]:
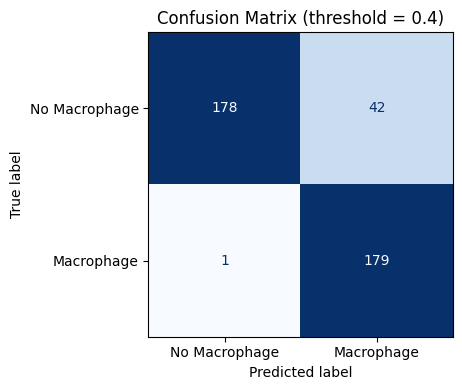
threshold_val = np.percentile(adata.obs['Macrophage_pred'], 50)
print(f"Median prediction score (threshold): {threshold_val:.4f}")
adata.obs['Macrophage_pred_binary'] = (adata.obs['Macrophage_pred'] >= 0.4).astype(int)
adata.obs['Macrophage_pred_binary'].value_counts()
Median prediction score (threshold): 0.4995
[28]:
Macrophage_pred_binary
1 221
0 179
Name: count, dtype: int64
[29]:
from sklearn.metrics import confusion_matrix, ConfusionMatrixDisplay
df_cm = adata.obs.dropna(subset=['Macrophage_pred_binary', 'Macrophage']).copy()
cm = confusion_matrix(df_cm['Macrophage'].astype(int),
df_cm['Macrophage_pred_binary'].astype(int))
disp = ConfusionMatrixDisplay(confusion_matrix=cm,
display_labels=['No Macrophage', 'Macrophage'])
fig, ax = plt.subplots(figsize=(5, 4))
disp.plot(ax=ax, colorbar=False, cmap='Blues')
ax.set_title('Confusion Matrix (threshold = 0.4)')
plt.tight_layout()
plt.show()
[30]:
n_cells = len(adata.obs)
marker_size = max(2, min(20, 2000 / n_cells)) # scale dot size to dataset
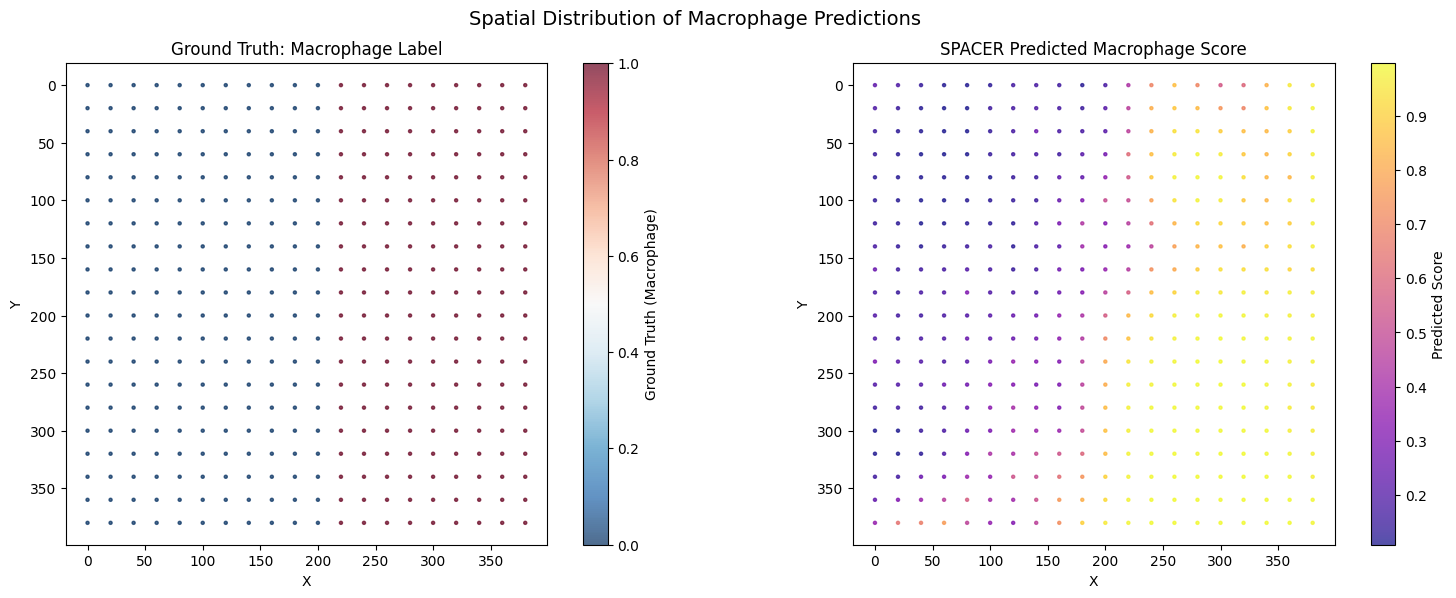
fig, axes = plt.subplots(1, 2, figsize=(16, 6))
sc0 = axes[0].scatter(
adata.obs['X'], adata.obs['Y'],
c=adata.obs['Macrophage'].astype(float),
cmap='RdBu_r', s=marker_size, alpha=0.7
)
plt.colorbar(sc0, ax=axes[0], label='Ground Truth (Macrophage)')
axes[0].set_title('Ground Truth: Macrophage Label')
axes[0].set_xlabel('X'); axes[0].set_ylabel('Y')
axes[0].invert_yaxis(); axes[0].set_aspect('equal')
vmin = adata.obs['Macrophage_pred'].quantile(0.01)
vmax = adata.obs['Macrophage_pred'].quantile(0.99)
sc1 = axes[1].scatter(
adata.obs['X'], adata.obs['Y'],
c=adata.obs['Macrophage_pred'],
cmap='plasma', s=marker_size, alpha=0.7, vmin=vmin, vmax=vmax
)
plt.colorbar(sc1, ax=axes[1], label='Predicted Score')
axes[1].set_title('SPACER Predicted Macrophage Score')
axes[1].set_xlabel('X'); axes[1].set_ylabel('Y')
axes[1].invert_yaxis(); axes[1].set_aspect('equal')
plt.suptitle('Spatial Distribution of Macrophage Predictions', fontsize=14)
plt.tight_layout()
plt.savefig('spatial_macrophage_predictions.png', dpi=150, bbox_inches='tight')
plt.show()
[31]:
matched = adata.obs[(adata.obs['Macrophage'] == 1) & (adata.obs['Macrophage_pred_binary'] == 1)]
print(f"True positives: {len(matched)} / {(adata.obs['Macrophage']==1).sum()} ground-truth positive cells")
matched[['X','Y','Macrophage','Macrophage_pred','Macrophage_pred_binary']].head(10)
True positives: 179 / 180 ground-truth positive cells
[31]:
| X | Y | Macrophage | Macrophage_pred | Macrophage_pred_binary | |
|---|---|---|---|---|---|
| CELL_00011-1 | 220.0 | 0.0 | 1 | 0.426641 | 1 |
| CELL_00012-1 | 240.0 | 0.0 | 1 | 0.695324 | 1 |
| CELL_00013-1 | 260.0 | 0.0 | 1 | 0.858642 | 1 |
| CELL_00014-1 | 280.0 | 0.0 | 1 | 0.699150 | 1 |
| CELL_00015-1 | 300.0 | 0.0 | 1 | 0.555146 | 1 |
| CELL_00016-1 | 320.0 | 0.0 | 1 | 0.600728 | 1 |
| CELL_00017-1 | 340.0 | 0.0 | 1 | 0.821454 | 1 |
| CELL_00018-1 | 360.0 | 0.0 | 1 | 0.956521 | 1 |
| CELL_00019-1 | 380.0 | 0.0 | 1 | 0.969552 | 1 |
| CELL_00031-1 | 220.0 | 20.0 | 1 | 0.463269 | 1 |
[ ]: