Spacer training Notebook
[1]:
# ── Example data paths (swap these for your own files) ──────────────────────
# Run `python create_example_data.py` once to generate the files below.
GENE_CSV = 'data/example_genes.csv' # reference gene list (377 genes)
ADATA_H5AD = 'data/example_spatial.h5ad' # 600 cells × 98 genes, spatial grid
SAMPLE_CSV = 'data/example_sample.csv' # multi-sample CSV (points at h5ad above)
IMMUNE_CELL = 'macrophage' # 'tcell' | 'macrophage' | 'bcell' etc.
# Synthetic label layout:
# T-cell infiltration → upper half (Y < 200)
# Macrophage infiltration → right half (X > 190)
#
# To train on real data, replace the three variables above and set
# immune_cell, radius, resolution in the BagsDataset call below.
[2]:
import os
import torch
import torch.nn as nn
import torch.optim as optim
import pandas as pd
import numpy as np
import scanpy as sc
from torch.utils.data import DataLoader, random_split
from sklearn.metrics import roc_auc_score
from tqdm import tqdm
from model.dataset import BagsDataset, custom_collate_fn
from model.model import MIL, EarlyStopping
[3]:
def load_all_genes(reference_gene_file):
all_genes = pd.read_csv(reference_gene_file)
return all_genes['Gene'].values.tolist()
Load reference gene list
all_genes = pd.read_csv(GENE_CSV) # swap GENE_CSV for ‘data/human_filtered.csv’ on real data all_genes
[4]:
all_genes = pd.read_csv(GENE_CSV)
all_genes
[4]:
| Gene | |
|---|---|
| 0 | TAP2 |
| 1 | IFI6 |
| 2 | TOP2A |
| 3 | PBK |
| 4 | TPX2 |
| ... | ... |
| 372 | GENE0295 |
| 373 | GENE0296 |
| 374 | GENE0297 |
| 375 | GENE0298 |
| 376 | GENE0299 |
377 rows × 1 columns
[5]:
all_genes = all_genes['Gene'].values.tolist()
── Option A: single AnnData file ───────────────────────────────────────────
adata = sc.read_h5ad(ADATA_H5AD) dataset = BagsDataset( adata, immune_cell=IMMUNE_CELL, radius=50, max_instances=500, n_genes=500, resolution=’low’, k=2, )
[ ]:
# Load dataset and create DataLoader (see the Data Preparation section for details)
adata = sc.read_h5ad(ADATA_H5AD)
dataset = BagsDataset(
adata,
immune_cell=IMMUNE_CELL,
radius=50,
max_instances=500,
n_genes=500,
resolution='low',
k=2, # Ensure 'k' matches the number of bags per batch
)
If you want to run the model for multiple datasets jointly
── Option B: multi-sample CSV ───────────────────────────────────────────────
adatas = pd.read_csv(SAMPLE_CSV) adatas # make sure columns are: adata, radius, resolution
[7]:
# ── Option B: multi-sample CSV ─────────────────────────────────────────────
adatas = pd.read_csv(SAMPLE_CSV)
adatas # make sure columns are: adata, radius, resolution
[7]:
| adata | radius | resolution | |
|---|---|---|---|
| 0 | data/example_spatial.h5ad | 50 | low |
| 1 | data/example_spatial.h5ad | 50 | low |
[8]:
dataset = BagsDataset(
SAMPLE_CSV,
immune_cell=IMMUNE_CELL,
max_instances=500,
n_genes=500,
k=2,
)
/work/DPDS/s439765/envs/spatial_tcr/lib/python3.9/site-packages/anndata/_core/anndata.py:1820: UserWarning: Variable names are not unique. To make them unique, call `.var_names_make_unique`.
utils.warn_names_duplicates("var")
/work/DPDS/s439765/envs/spatial_tcr/lib/python3.9/site-packages/anndata/_core/anndata.py:1820: UserWarning: Variable names are not unique. To make them unique, call `.var_names_make_unique`.
utils.warn_names_duplicates("var")
/work/DPDS/s439765/envs/spatial_tcr/lib/python3.9/site-packages/anndata/_core/anndata.py:1820: UserWarning: Variable names are not unique. To make them unique, call `.var_names_make_unique`.
utils.warn_names_duplicates("var")
/work/DPDS/s439765/envs/spatial_tcr/lib/python3.9/site-packages/anndata/_core/anndata.py:1820: UserWarning: Variable names are not unique. To make them unique, call `.var_names_make_unique`.
utils.warn_names_duplicates("var")
Immune cell: Macrophage
Reading adata from data/example_spatial.h5ad
[1 0]
Tumor cells shape after filtering: (400, 98)
Selecting top 500 genes based on mean expression
tumor_cells.obs[Macrophage] is already binary. Skipping binarization.
Processing: adata=example_spatial.h5ad, radius=50, resolution=low
Reading adata from data/example_spatial.h5ad
[1 0]
Tumor cells shape after filtering: (400, 98)
Selecting top 500 genes based on mean expression
tumor_cells.obs[Macrophage] is already binary. Skipping binarization.
Processing: adata=example_spatial.h5ad, radius=50, resolution=low
Creating Bags with radius 50: 100%|████████████████████████████| 600/600 [00:00<00:00, 13481.96it/s]
Creating Bags with radius 50: 100%|████████████████████████████| 600/600 [00:00<00:00, 13821.22it/s]
Total batches created: 360
[9]:
device = torch.device("cuda" if torch.cuda.is_available() else "cpu")
if device.type == "cuda":
print(f"Using device: {device} ({torch.cuda.get_device_name(torch.cuda.current_device())})")
else:
print(f"Using device: {device}")
print("=====================================")
Using device: cpu
=====================================
[10]:
model = MIL(all_genes).to(device)
criterion = nn.BCELoss().to(device)
optimizer = optim.AdamW(model.parameters(), lr=0.01, weight_decay=0.02)
early_stopping = EarlyStopping(patience=5, delta=0.001)
[11]:
output_dir = 'sample_output'
os.makedirs(output_dir, exist_ok=True)
[12]:
train_size = int(0.9 * len(dataset))
val_size = len(dataset) - train_size
train_dataset, val_dataset = random_split(dataset, [train_size, val_size])
train_loader = DataLoader(train_dataset, batch_size=1, shuffle=True, collate_fn=custom_collate_fn)
val_loader = DataLoader(val_dataset, batch_size=1, shuffle=True, collate_fn=custom_collate_fn)
[13]:
best_val_loss = float('inf')
best_model_path = os.path.join(output_dir, 'best_model.pth')
# Save spacer scores before training
spacer_scores_before_training = model.immunogenicity.ig.clone().detach().cpu()
spacer_scores_before_training = [score.item() for score in spacer_scores_before_training]
[14]:
def save_metrics(epoch, train_loss, val_loss, val_auroc, a, b, alpha, beta, output_dir):
file_path = os.path.join(output_dir, 'training_metrics.csv')
if not os.path.exists(file_path):
# Create the CSV file with headers
with open(file_path, 'w') as f:
f.write('Epoch,Train Loss,Val Loss,Val AUROC,a,b,alpha,beta\n')
# Append metrics for the current epoch
with open(file_path, 'a') as f:
f.write(f'{epoch},{train_loss},{val_loss},{val_auroc},{a},{b},{alpha},{beta}\n')
def save_spacer_scores(epoch, all_genes, spacer_scores_before_training, spacer_scores_after_training, output_dir):
# Create a DataFrame with IG scores before and after the current epoch
spacer_score_data = {
'Gene': all_genes,
'SPACER Score Before Training': spacer_scores_before_training,
'SPACER Score After Training': spacer_scores_after_training,
}
df = pd.DataFrame(spacer_score_data)
# Calculate the difference and add it as a new column
df['Difference'] = df['SPACER Score After Training'] - df['SPACER Score Before Training']
df = df.sort_values(by='Difference', ascending=False)
# Save to a CSV file for each epoch
output_path = os.path.join(output_dir, f'spacer_score_changes_epoch_{epoch+1}.csv')
df.to_csv(output_path, index=False)
Training
[15]:
num_epochs = 2
selection = 'positive' # Choose 'positive(induce)' or 'negative(repel)' based on your research focus
[16]:
for epoch in range(num_epochs):
model.train()
running_loss = 0.0
# Lists to store outputs and labels for AUROC calculation
all_outputs = []
all_labels = []
with tqdm(train_loader, unit="batch") as tepoch:
for i, batch_data in enumerate(tepoch):
tepoch.set_description(f"Epoch {epoch+1}/{num_epochs}")
optimizer.zero_grad()
# Unpack the batch data
distances_list, gene_expressions_list, labels_list, core_idxs_list, gene_names_list, cell_ids_list = batch_data
# Move data to device and prepare labels
distances_list = [distances.to(device) for distances in distances_list]
gene_expressions_list = [gene_exp.to(device) for gene_exp in gene_expressions_list]
labels = torch.stack(labels_list).float().to(device)
current_genes_list = gene_names_list # List of gene names for each bag
# Forward pass
outputs = model(distances_list, gene_expressions_list, current_genes_list)
if outputs is None:
continue # Skip this batch if the model returns None
if outputs.shape[0] != labels.shape[0]:
# Handle mismatch in batch sizes if necessary
continue
# Compute BCE loss
if selection == 'negative':
labels = 1 - labels
loss = criterion(outputs, labels)
loss.backward()
optimizer.step()
running_loss += loss.item()
tepoch.set_postfix(loss=loss.item())
# Accumulate outputs and labels for AUROC calculation
all_outputs.extend(outputs.detach().cpu().numpy())
all_labels.extend(labels.cpu().numpy())
train_loss = running_loss / len(train_loader)
# Compute Training AUROC
try:
epoch_auc = roc_auc_score(all_labels, all_outputs)
except ValueError:
epoch_auc = float('nan') # Handle case where AUROC can't be computed
print(f'Epoch [{epoch+1}/{num_epochs}], Loss: {train_loss:.4f}, AUROC: {epoch_auc:.4f}')
# Validation phase
model.eval()
val_loss = 0.0
val_all_outputs = []
val_all_labels = []
with torch.no_grad():
with tqdm(val_loader, unit="batch") as vtepoch:
for val_batch_data in vtepoch:
# Unpack validation batch data
val_distances_list, val_gene_expressions_list, val_labels_list, val_core_idxs_list, val_gene_names_list, val_cell_ids_list = val_batch_data
# Move data to device and prepare labels
val_distances_list = [distances.to(device) for distances in val_distances_list]
val_gene_expressions_list = [gene_exp.to(device) for gene_exp in val_gene_expressions_list]
val_labels = torch.stack(val_labels_list).float().to(device)
val_current_genes_list = val_gene_names_list # List of gene names for each bag
# Forward pass
val_outputs = model(val_distances_list, val_gene_expressions_list, val_current_genes_list)
if val_outputs is None:
continue # Skip this batch if the model returns None
if val_outputs.shape[0] != val_labels.shape[0]:
# Handle mismatch in batch sizes if necessary
continue
# Compute BCE loss
if selection == 'negative':
val_labels = 1 - val_labels
loss = criterion(val_outputs, val_labels)
val_loss += loss.item()
vtepoch.set_postfix(val_loss=loss.item())
# Accumulate outputs and labels for AUROC calculation
val_all_outputs.extend(val_outputs.detach().cpu().numpy())
val_all_labels.extend(val_labels.cpu().numpy())
val_loss /= len(val_loader)
# Compute Validation AUROC
try:
val_epoch_auc = roc_auc_score(val_all_labels, val_all_outputs)
except ValueError:
val_epoch_auc = float('nan') # Handle case where AUROC can't be computed
print(f'Validation Loss: {val_loss:.4f}, Validation AUROC: {val_epoch_auc:.4f}')
# Save the best model
if val_loss < best_val_loss:
best_val_loss = val_loss
torch.save(model.state_dict(), best_model_path)
print(f"Best model saved with validation loss {val_loss:.4f}")
torch.save(model.state_dict(), os.path.join(output_dir, f'model_epoch_{epoch+1}.pth'))
a = model.distance.a.clone().detach().cpu().numpy()
b = model.gene_expression.b.clone().detach().cpu()
alpha = model.alpha.clone().detach().cpu()
beta = model.beta.clone().detach().cpu()
# Save metrics
save_metrics(epoch+1, train_loss, val_loss, val_epoch_auc,a,b,alpha,beta, output_dir)
# Save SPACER scores after each epoch
spacer_scores_after_training = model.immunogenicity.ig.clone().detach().cpu()
spacer_scores_after_training = [score.item() for score in spacer_scores_after_training]
save_spacer_scores(epoch, all_genes, spacer_scores_before_training, spacer_scores_after_training, output_dir)
Epoch 1/2: 100%|██████████| 324/324 [00:00<00:00, 416.36batch/s, loss=0.462]
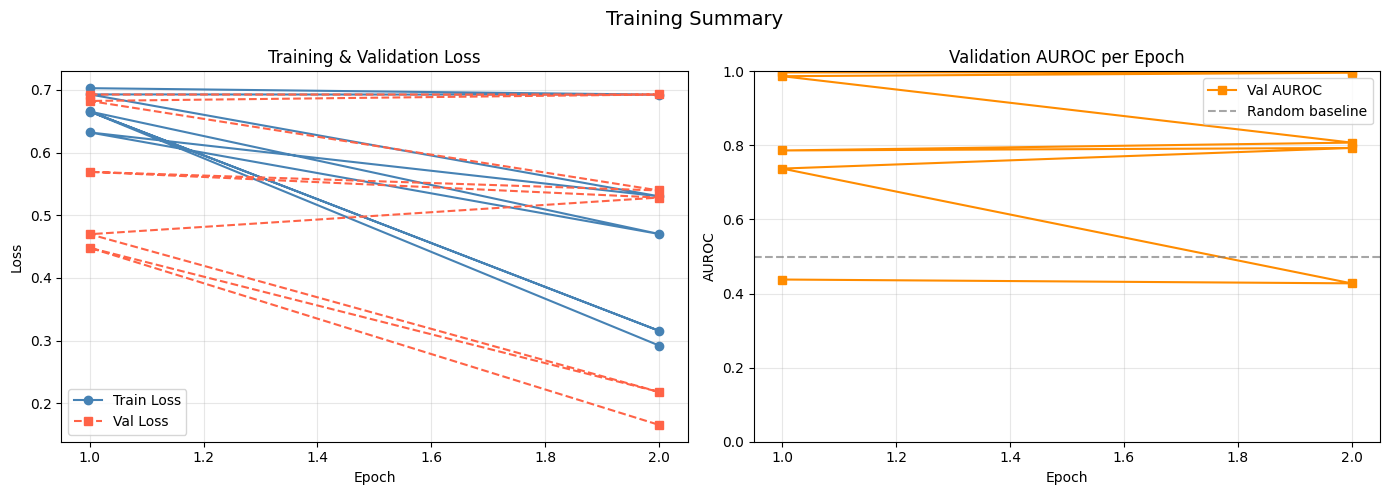
Epoch [1/2], Loss: 0.6657, AUROC: 0.7575
100%|██████████| 36/36 [00:00<00:00, 986.86batch/s, val_loss=0.399]
Validation Loss: 0.4481, Validation AUROC: 0.9977
Best model saved with validation loss 0.4481
Epoch 2/2: 100%|██████████| 324/324 [00:00<00:00, 449.16batch/s, loss=0.115]
Epoch [2/2], Loss: 0.2922, AUROC: 0.9920
100%|██████████| 36/36 [00:00<00:00, 1078.31batch/s, val_loss=0.0617]
Validation Loss: 0.1656, Validation AUROC: 1.0000
Best model saved with validation loss 0.1656
Training Results Visualization
[17]:
import matplotlib.pyplot as plt
# Training & validation loss / AUROC curves
metrics_df = pd.read_csv(os.path.join(output_dir, 'training_metrics.csv'))
fig, axes = plt.subplots(1, 2, figsize=(14, 5))
axes[0].plot(metrics_df['Epoch'], metrics_df['Train Loss'],
marker='o', label='Train Loss', color='steelblue')
axes[0].plot(metrics_df['Epoch'], metrics_df['Val Loss'],
marker='s', linestyle='--', label='Val Loss', color='tomato')
axes[0].set_xlabel('Epoch')
axes[0].set_ylabel('Loss')
axes[0].set_title('Training & Validation Loss')
axes[0].legend()
axes[0].grid(True, alpha=0.3)
axes[1].plot(metrics_df['Epoch'], metrics_df['Val AUROC'],
marker='s', color='darkorange', label='Val AUROC')
axes[1].axhline(y=0.5, color='gray', linestyle='--', alpha=0.7, label='Random baseline')
axes[1].set_ylim(0, 1)
axes[1].set_xlabel('Epoch')
axes[1].set_ylabel('AUROC')
axes[1].set_title('Validation AUROC per Epoch')
axes[1].legend()
axes[1].grid(True, alpha=0.3)
plt.suptitle('Training Summary', fontsize=14)
plt.tight_layout()
plt.savefig(os.path.join(output_dir, 'training_curves.png'), dpi=150, bbox_inches='tight')
plt.show()
[18]:
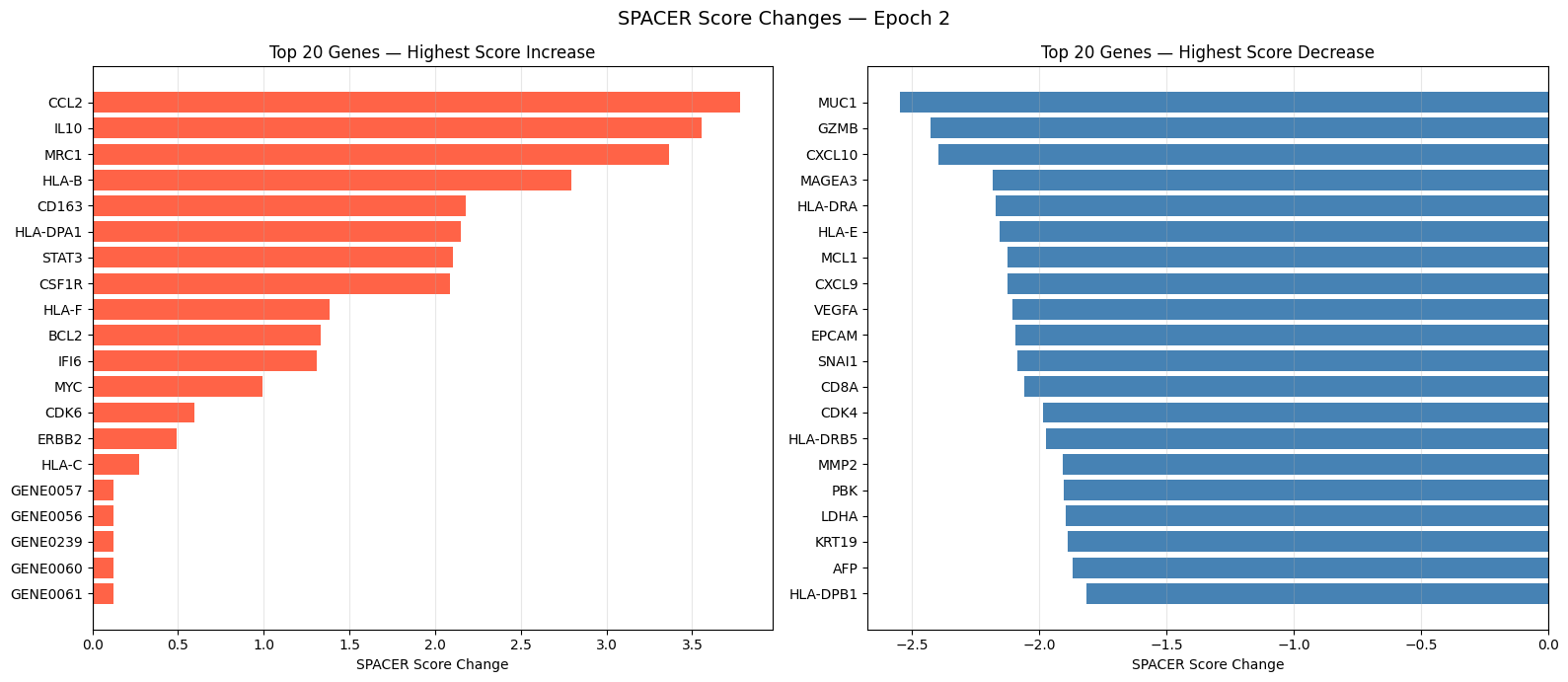
# Top genes by SPACER score change (last epoch)
import glob
spacer_files = sorted(glob.glob(os.path.join(output_dir, 'spacer_score_changes_epoch_*.csv')))
if spacer_files:
last_df = pd.read_csv(spacer_files[-1]).sort_values('Difference', ascending=False)
top_n = 20
top_up = last_df.head(top_n)
top_dn = last_df.tail(top_n).sort_values('Difference', ascending=True)
fig, axes = plt.subplots(1, 2, figsize=(16, 7))
axes[0].barh(top_up['Gene'], top_up['Difference'], color='tomato')
axes[0].axvline(x=0, color='black', linewidth=0.8)
axes[0].set_xlabel('SPACER Score Change')
axes[0].set_title(f'Top {top_n} Genes — Highest Score Increase')
axes[0].invert_yaxis()
axes[0].grid(True, axis='x', alpha=0.3)
axes[1].barh(top_dn['Gene'], top_dn['Difference'], color='steelblue')
axes[1].axvline(x=0, color='black', linewidth=0.8)
axes[1].set_xlabel('SPACER Score Change')
axes[1].set_title(f'Top {top_n} Genes — Highest Score Decrease')
axes[1].invert_yaxis()
axes[1].grid(True, axis='x', alpha=0.3)
plt.suptitle(f'SPACER Score Changes — Epoch {len(spacer_files)}', fontsize=14)
plt.tight_layout()
plt.savefig(os.path.join(output_dir, 'spacer_score_changes.png'), dpi=150, bbox_inches='tight')
plt.show()
else:
print("No SPACER score files found in output_dir.")
[19]:
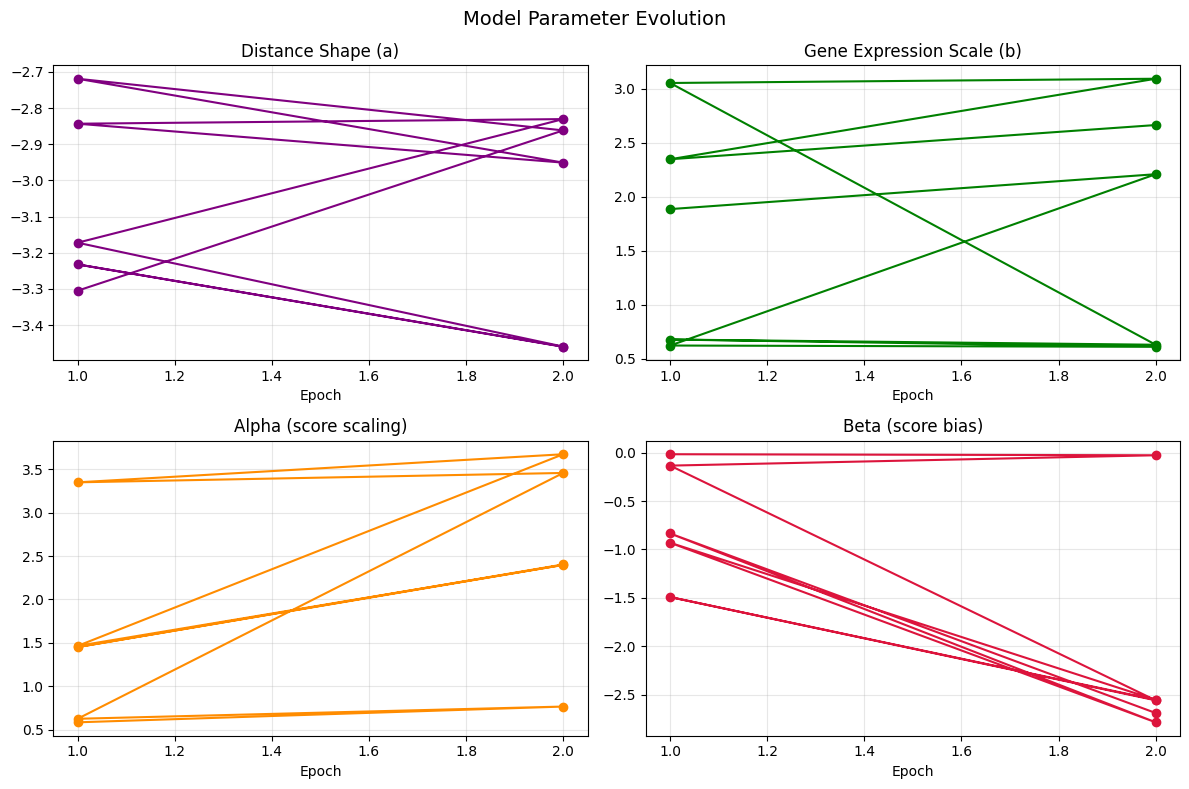
# Model parameter evolution across epochs
def _parse_param(val):
s = str(val).strip().strip('[]')
try:
return float(s)
except ValueError:
return float('nan')
fig, axes = plt.subplots(2, 2, figsize=(12, 8))
param_cfg = {
'a': ('Distance Shape (a)', 'purple'),
'b': ('Gene Expression Scale (b)', 'green'),
'alpha': ('Alpha (score scaling)', 'darkorange'),
'beta': ('Beta (score bias)', 'crimson'),
}
for ax, (col, (title, color)) in zip(axes.ravel(), param_cfg.items()):
vals = metrics_df[col].apply(_parse_param)
ax.plot(metrics_df['Epoch'], vals, marker='o', color=color)
ax.set_xlabel('Epoch')
ax.set_title(title)
ax.grid(True, alpha=0.3)
plt.suptitle('Model Parameter Evolution', fontsize=14)
plt.tight_layout()
plt.savefig(os.path.join(output_dir, 'parameter_evolution.png'), dpi=150, bbox_inches='tight')
plt.show()
[ ]: